
El Laboratorio de Genómica de la RAI provee a la comunidad científica una amplia gama de servicios que facilitan la investigación genómica con un abordaje funcional. Las herramientas genómicas que el laboratorio ofrece están dirigidas hacia aplicaciones de secuenciación de genoma completo, secuenciación de transcriptoma y análisis de expresión, secuenciación dirigida a regiones blanco y secuenciación de exoma, entre otras, mediante el uso de las plataformas de Ilumina MiSeq y HiSeq2500. Ofrecemos soluciones flexibles para una gran variedad de aplicaciones, tales como las que se enlistan a continuación:
- Caracterización y análisis de las variaciones genéticas en el genoma humano completo (secuenciación de genoma completo)
- Caracterización y análisis de las variaciones en regiones exónicas (secuenciación de exoma)
- Secuenciación de novo de genomas de cualquier tamaño
- Identificación de variaciones en regiones blanco del genoma
- Diseño y análisis de páneles de variantes específicas
- Composición de la microbiota (metagenómica)
- Análisis de variaciones en DNA mitocondrial humano
- Análisis de la expresión génica del transcriptoma humano (secuenciación del transcriptoma completo)
- Identificación de RNA no codificantes con efecto regulador
- Análisis de expresión de genes blanco involucrados en vías de señalización específicas
- Descubrimiento e identificación de RNA pequeños
- Determinación de biomarcadores específicos
Visite Fundamentos para conocer los aspectos técnicos de estas plataformas. Si desea saber más sobre Illumina® y sus productos visite la página www.illumina.com.
Fundamento de las plataformas
El proceso de secuenciación está dividido en 3 etapas:
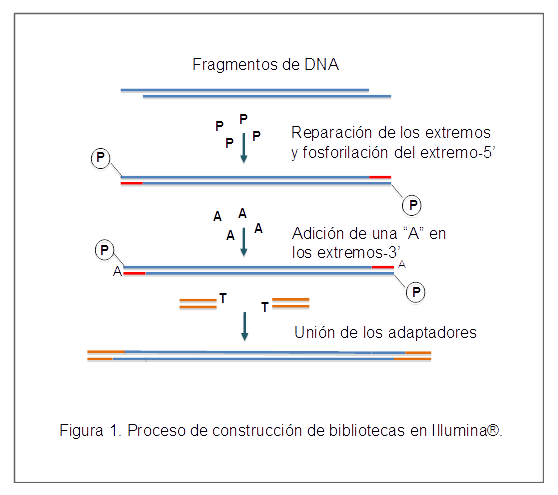
Preparación de bibliotecas: El objetivo de la preparación de la biblioteca o preparación de muestras es obtener fragmentos de las muestras de un tamaño específico con adaptadores unidos en ambos extremos (Figura 1).
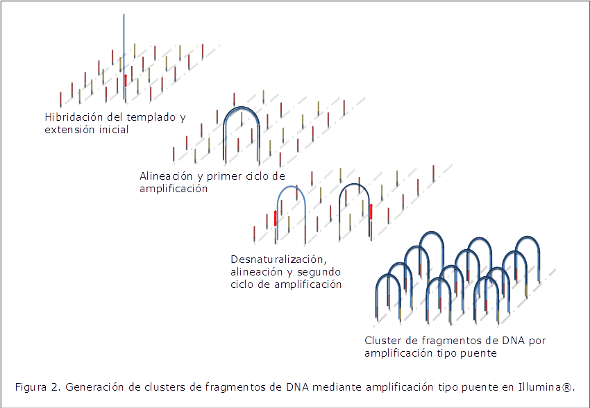
Generación de clusters: Durante la generación de clusters las muestras se hibridan a la superficie de la celda de flujo a través de los adaptadores previamente unidos, posteriormente se extienden por una amplificación tipo puente y se linearizan. La celda de flujo contiene los oligonucleótidos (primers) de amplificación complementarios a los adaptadores unidos a las muestras fragmentadas. Los fragmentos se unen a los primers por complementariedad y estos son extendidos por la polimerasa, generándose una copia complementaria de la cadena original. La molécula de doble cadena se desnaturaliza y el templado original se desecha, quedando la nueva hebra de DNA sintetizado unido covalentemente a la superficie de la celda en uno de sus extremos. Posteriormente para la amplificación, el extremo de la hebra sencilla que ha quedado libre hibrida con un oligonucleótido adyacente localizado sobre la superficie de la celda para formar un puente y el primer es entonces extendido por la polimerasa para generar un puente de doble cadena. El puente es desnaturalizado, resultando en dos copias de cadena sencilla del templado, unidas covalentemente a la superficie. Los ciclos de amplificación se repiten hasta formar múltiples puentes.
Finalmente, los puentes de doble cadena se desnaturalizan, se bloquean los extremos 3’ de las hebras de DNA con dNTPs con el fin de evitar que cualquier DNA o nucleótido pueda interferir durante la secuenciación y se hibridan los primers de secuenciación (Figura 2).
-
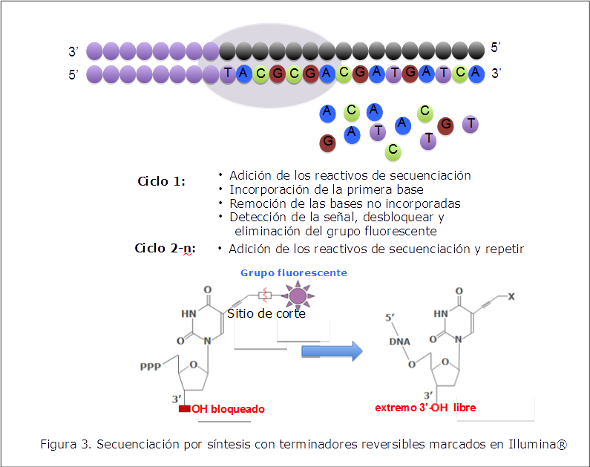
Secuenciación: La tecnología de secuenciación de Illumina está basada en el procedimiento de secuenciación por síntesis (SBS) con terminadores reversibles marcados. Los cuatro nucleótidos identificados con una marca o grupo fluorescente diferente se adicionan en cada ciclo durante todo el proceso de secuenciación. La secuenciación se lleva a cabo a través de reacciones de adición de un solo nucleótido por ciclo, ya que estos contienen en la posición 3’-OH un grupo que bloquea y previene que la polimerasa incorpore bases adicionales. En cada ciclo se produce una serie de eventos que ocurren en el siguiente orden: a) Un nucleótido es incorporado por la polimerasa; b) los nucleótidos no incorporados son lavados y retirados; c) se captura una imagen de la celda con el fin de identificar la señal de fluorescencia emitida y por lo tanto el nucleótido incorporado; d) los grupos fluorescentes son liberados; y e) el nucleótido 3’-OH es químicamente desbloqueado. Para leer el extremo opuesto de cada fragmento (paired end), las hebras se desnaturalizan, se remueve la hebra recién sintetizada y se regeneran los clusters a través de la reamplificación por puentes. Posteriormente se hibridan los extremos con el primer reverso y el proceso de secuenciación procede como se describió anteriormente (Figura 3).
Equipos
| Manual de procedimientos del MiSeq
Guía de usuario |
MiSeq Desktop Sequencer |

| Manual del usuario (Illumina)
Guía de usuario |
cBot System |

| Manual del espectrofotómetro NanoDrop 2000/2000c
Guía de usuario |
NanoDrop Spectrophotometer |

| Manual del bioanalizador 2100 (Expert User's Guide)
Bioanalyzer Maintenance and Troubleshooting Guide Agilent guía del kit de ARN (6000 Nano) Guía de usuario |
Agilent 2100 Bioanalyzer |
| Guía de usuario del Sonicador Covaris 2200 | Sonicador Covaris 2200 |
| Manual del Qubit 3.0 Fluorometer (User Guide)
Guía de usuario |
Qubit 3.0 Fluorometer |
| Guía de usuario de la campana de flujo laminar | Campana de flujo laminar |
Investigación
Laboratorio de Neuroendocrinología y Genómica
Dra. Laura C. Hernández Ramírez,
Investigadora Principal.
Dra. Rosa G. Rebollar Vega,
Técnica Académica.
Nuestro laboratorio investiga las causas genéticas de los tumores neuroendocrinos de la glándula hipófisis y otros órganos y sus relaciones con los fenotipos observados en la clínica utilizando técnicas de secuenciación de nueva generación. A la par de determinar el tipo y frecuencia de defectos genéticos que subyacen estas neoplasias en nuestra población, buscamos identificar nuevas asociaciones patogénicas y marcadores de comportamiento clínico. Por otro lado, estudiamos las consecuencias moleculares de dichas alteraciones sobre los tejidos afectados por medio de modelos celulares y murinos. Finalmente, analizamos el potencial patogénico de variantes génicas para sustentar su clasificación clínica utilizando herramientas in silico y experimentos de validación funcional in vitro
- Bases genéticas y moleculares de los tumores neuroendocrinos hipofisarios (financiado con fondos de la Coordinación de la Investigación Científica, CIC, Universidad Nacional Autónoma de México, UNAM).
- Evaluación de marcadores genéticos y moleculares como herramientas de pronóstico del comportamiento clínico en la enfermedad de Cushing (financiado con fondos de la CIC, UNAM).
- Detección, análisis y validación funcional de variantes genéticas germinales causales de neoplasias neuroendocrinas en pacientes mestizos mexicanos (financiado con fondos del Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica, UNAM).
- Repertorio de fenotipos clínicos y defectos genéticos subyacentes en pacientes mexicanos con síndromes de neoplasia endocrina múltiple y neoplasias neuroendocrinas aisladas familiares (financiado con fondos de la Sociedad Mexicana de Nutrición y Endocrinología).
Coordinación de la Investigación Científica,
Universidad Nacional Autónoma de México

Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica (PAPIIT), Dirección General de Asuntos del Personal Académico, Universidad Nacional Autónoma de México.

Programa de financiamiento a proyectos de investigación, Sociedad Mexicana de Nutrición y Endocrinología.

https://endocrinologia.org.mx/indexdres.php
Equipment Grant, Society for Endocrinology.

Contacto
Para mayor información o asesoría sobre las plataformas favor de contactar a:
-
Dra. Laura Cristina Hernández Ramírez
Investigadora responsable
- laura.hernandezcic.unam.mx
- +01 (55) 5487 0900 Ext. 6328
-
M. en C. Rosa Gloria Rebollar Vega
Técnica Académica
- rebollarcic.unam.mx
- +01 (55) 5487 0900 Ext. 6328 ó 6329